Introduction: Stability in Standards, Evolution in Regulation
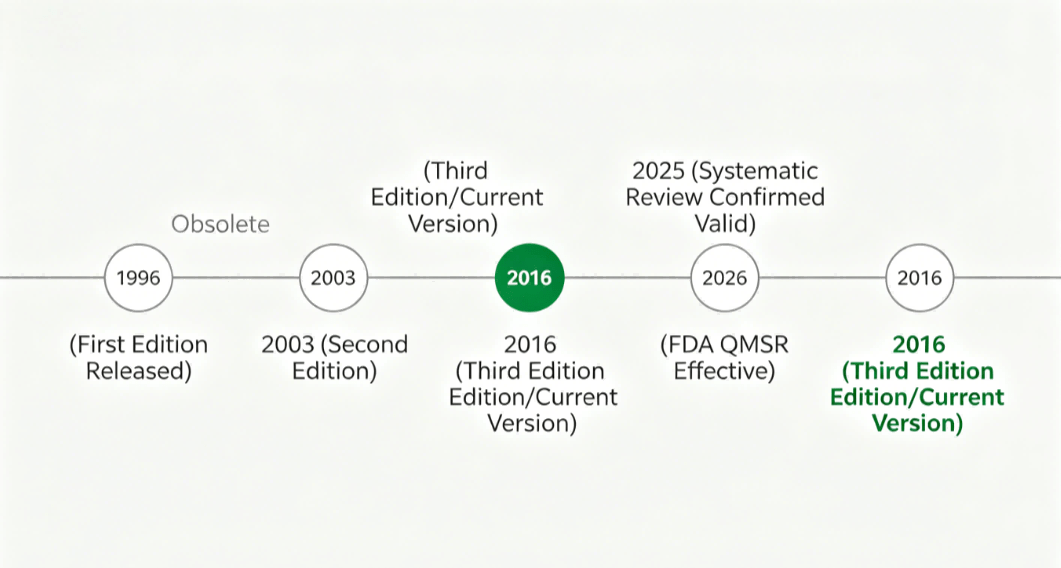
For medical device manufacturers and their supply chain partners, 2025 and 2026 have been pivotal years. The ISO 13485 standard — the globally recognized framework for medical device quality management systems — underwent its periodic systematic review and was confirmed as current and fit for purpose, meaning no revision will take place at this time. The 2016 version remains the current mainstream standard for the global medical device industry.
However, while the standard itself remains unchanged, the regulatory environment surrounding it has evolved significantly. The most consequential development: the U.S. Food and Drug Administration’s Quality Management System Regulation (QMSR) officially took effect on February 2, 2026, incorporating ISO 13485:2016 by reference into U.S. federal law for the first time.
This article examines the current status of ISO 13485, the QMSR transition, and what these developments mean for medical device PCBA manufacturing.
At Guangzhou Huachuang Precision Technology Co., Ltd. (HCJMPCBA), we maintain ISO9001 quality management and ISO13485 medical device quality management certifications across our nearly 3,500 m² manufacturing facility. Our multiple high-speed SMT lines and comprehensive quality systems support medical device PCBA from prototype to volume production.
ISO 13485:2016 — Reaffirmed, Not Revised
The Systematic Review Process
ISO standards undergo systematic reviews at regular intervals to ensure they remain relevant and effective. ISO 13485:2016 entered its five-year review cycle in 2024, with feedback from ISO member bodies collected through mid-2025.
The outcome: ISO 13485:2016 has been confirmed and will remain the current version for the foreseeable future. The standard’s framework continues to serve its purpose — ensuring consistent, safe, and effective medical devices through a process-based approach to quality management.
What Reaffirmation Means for Manufacturers
For organizations already certified to ISO 13485:2016, the reaffirmation provides practical reassurance:
-
Continuity — Certification schemes, audits, and supplier requirements continue under familiar expectations
-
Stability — The foundation on which medical device QMSs are built remains stable
-
Confidence — The reaffirmation reflects global consensus that the standard’s framework remains robust
However, reaffirmation does not mean the environment around ISO 13485 has stopped evolving. Regulators worldwide are adapting to technological change, particularly in digital health, AI-enabled systems, and software lifecycle management.
Iso 13485 Standard Evolution Timeline
The FDA QMSR: A Historic Alignment
From QSR to QMSR
The FDA’s transition from the legacy Quality System Regulation (QSR — 21 CFR Part 820) to the Quality Management System Regulation (QMSR) represents the most significant U.S. medtech regulatory change in nearly 30 years.
Key elements of the QMSR:
-
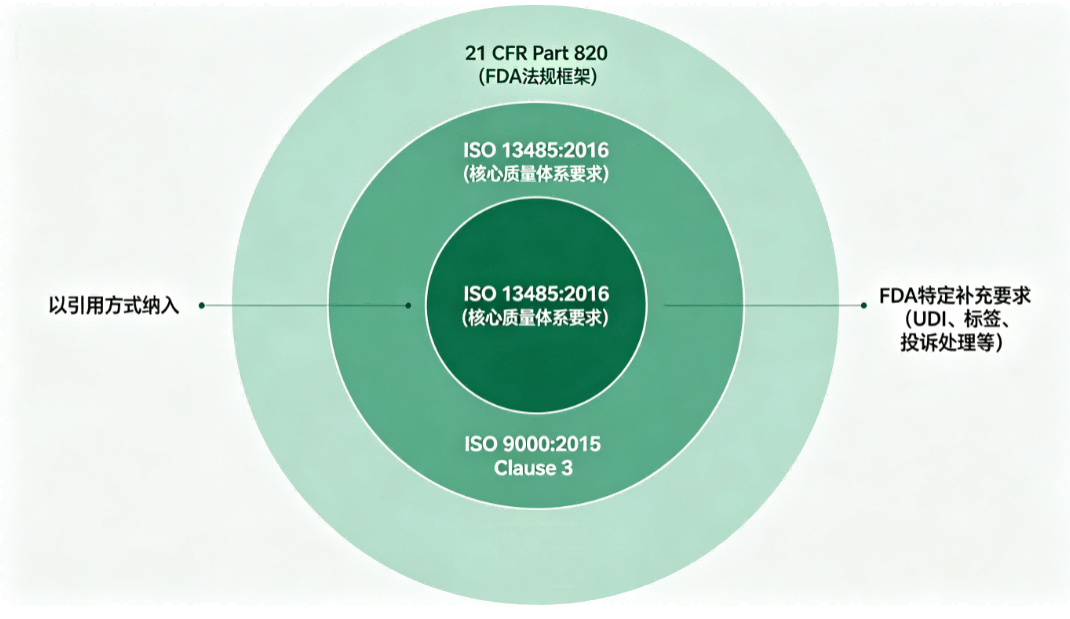
ISO 13485:2016 is now the primary quality management system requirement within Part 820
-
FDA incorporates Clause 3 of ISO 9000:2015 for aligned terminology
-
Part 820 is restructured to function as an overlay on ISO 13485, with FDA-specific provisions retained where U.S. regulatory obligations require additional clarity
The End of Dual Compliance
Before QMSR, U.S. manufacturers followed FDA’s QSR while the rest of the world relied on ISO 13485. This dual system forced companies to maintain two parallel quality frameworks, duplicate documentation, and pass separate audits.
Industry surveys from 2025 showed that 78% of U.S. medtech firms operated separate QSR and ISO 13485 systems, with an average annual compliance cost of $120,000–$180,000 per facility to maintain both.
QMSR eliminates this friction by harmonizing U.S. rules with ISO 13485:2016. Early analysis estimates that fully harmonized companies can reduce compliance costs by 30–40% and cut audit preparation time by half.
FDA-Specific Provisions Retained
While QMSR adopts ISO 13485 as the core framework, FDA retains or adds specific provisions where U.S. regulatory obligations drive additional clarity:
-
Records and traceability — Explicitly linked to U.S. requirements for complaint evaluation, servicing records, and medical device reporting
-
Labeling and packaging controls — Carried forward in Part 820
-
UDI requirements — Captured in records with data flow from labeling to production and postmarket processes
FDA has stated it will have the authority to inspect management review, internal quality audit, and supplier audit reports.
Fda Qmsr Compliance Framework Structure Diagram
New Inspection Approach Under QMSR
From QSIT to Risk-Based Inspections
The QMSR transition brought a fundamental change in how FDA conducts medical device inspections. On January 30, 2026 — just three days before QMSR became effective — FDA released a new compliance program manual: “Inspection of Medical Device Manufacturers” (CP 7382.850).
This replaces the longstanding Quality System Inspection Technique (QSIT) and establishes the agency’s inspectional playbook under QMSR.
What Inspectors Now Evaluate
Under QMSR, FDA inspections now evaluate quality systems using the structure and terminology of ISO 13485:
-
Quality manuals, SOPs, management reviews, and CAPA systems must reflect ISO 13485 alignment
-
Inspectors select elements based on firm-specific risks, rather than standardized sampling
-
The shift from QSIT to ISO-aligned, risk-based inspections means investigators now focus on areas most relevant to each manufacturer’s specific operations
Early FDA inspection data (February 2 – April 20, 2026) showed that 51.2% of QMSR inspections cited inadequate risk integration as a top finding, compared to 32% pre-QMSR.

Comparison Chart Of Fda Inspection Method Evolution
ISO 23485: New Guidance Under Development
Recognizing the need for stronger implementation support, work is underway on ISO 23485 — a new guidance document for the application of ISO 13485:2016.
Key facts about ISO 23485:
-
Approved for development in January 2025
-
Currently at “Close of comment period” stage (20.60)
-
Will provide guidance on the intent of requirements in ISO 13485:2016 with examples of possible steps organizations can take to meet requirements
-
Particularly relevant for emerging areas such as Software as a Medical Device (SaMD)
The reaffirmation of ISO 13485 and the creation of new guidance are complementary developments — the reaffirmation preserves a stable foundation; the guidance ensures that foundation remains relevant in a rapidly changing regulatory world.
What This Means for Medical Device PCBA Supply Chains
For hardware engineers and procurement teams sourcing PCBA for medical devices, these regulatory developments have practical implications:
1. Supply Chain Quality Requirements Are Converging
With FDA aligning with ISO 13485, the quality requirements for medical device PCBA are becoming more consistent across markets. A PCBA supplier certified to ISO 13485 now meets the core quality framework for both U.S. and international markets.
2. Documentation and Traceability Are Paramount
Under QMSR, FDA inspectors may request management review, internal audit, and supplier audit reports. For PCBA suppliers, this means:
-
Complete material traceability is essential — not optional
-
Audit records must be maintained and readily retrievable
-
Supplier oversight documentation must be comprehensive
3. Risk Management Must Be Continuous
Early QMSR inspection data shows risk integration is a top area of focus. PCBA suppliers must demonstrate risk-based thinking throughout the manufacturing process, not just at design phase.
4. Global Harmonization Is Accelerating
The EU, UK, Singapore, and other markets are moving in the same direction through frameworks like MDSAP (Medical Device Single Audit Program). A supplier already certified to ISO 13485 is positioned to serve multiple regulated markets efficiently.
HCJMPCBA: ISO13485-Certified Medical Device PCBA Manufacturing
At Guangzhou Huachuang Precision Technology Co., Ltd. (HCJMPCBA), we have maintained ISO13485 medical device quality management certification as a core part of our manufacturing capability. Our nearly 3,500 m² ESD‑protected facility and multiple high-speed SMT lines operate under documented quality systems that meet the requirements of medical device manufacturers.
Our ISO13485-certified capabilities include:
-
Full material traceability — Every component tracked from receipt through assembly to delivery via our MES system
-
Documented process controls — All manufacturing processes validated and monitored
-
Comprehensive inspection — 3D SPI, AOI, X-ray, ICT, and functional testing integrated into production flow
-
Supplier quality management — Formal supplier evaluation and oversight processes
-
Audit-ready documentation — Complete quality records maintained and retrievable
-
Scalable production — From prototypes to volume production with consistent quality standards
We serve medical device customers across diagnostic equipment, patient monitoring, implantable devices, and laboratory instrumentation — applications where quality and traceability are not optional.
Guangzhou Huachuang Precision Technology Co., Ltd. (HCJMPCBA) — ISO13485-certified PCBA manufacturing for the medical device industry.